Development of rapid real-time RT-PCR (ƩS COVID-19)
A newly developed one-step quadruplex real-time RT-PCR assay was designed to target ORF1ab, ORF3a, and N genes of SARS-CoV-2, and ASBVd as an internal control (Fig. 2). The initial setting used the standard protocol as described in the previous section. The total run time was 67 min 56 s.

The SARS-CoV-2 genome was targeted by three sets of primers and probes specific for ORF1ab, ORF3a, and N genes. Red half arrows represented forward and reverse primers. Green half arrow represented probes. The figure was created with BioRender.com.
To develop a rapid real-time RT-PCR assay, the protocol was first adjusted to a 5 min RT step followed by 1 min initial denaturation and 40 cycles of 2 s denaturation at 92 °C and 4 s annealing/extension at 60 °C. In addition, the fast mode of QuantStudio 5 which automatically adjusted the ramp rate from 1.6 °C/s in standard mode to 4.13 °C/s denaturation and 3.16 °C/s annealing was also applied. The performance of this adjusted setting was comparable with the initial setting as shown in Fig. 3.
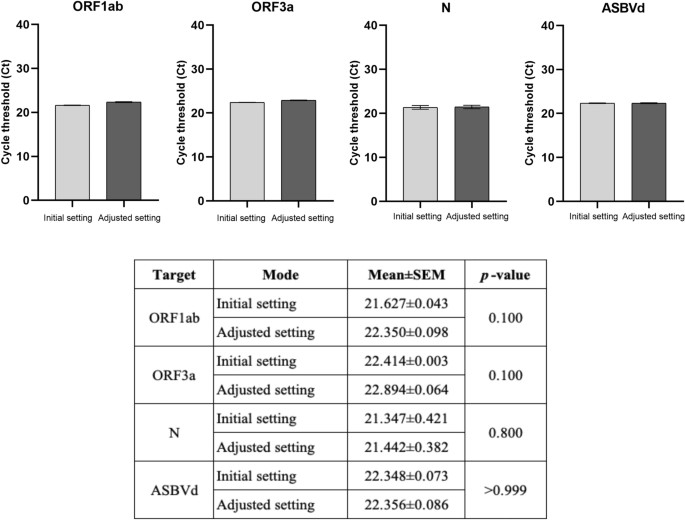
The performance of the real-time RT-PCR assay in the adjusted setting compared with the initial setting (the standard protocol). The mean ± SEM cycle threshold (Ct) values from an experiment with triplicate amplification were compared between two settings in each SARS-CoV-2 gene target (ORF1ab, ORF3a, and N genes) and the internal control (ASBVd). An unpaired t-test was used for comparison analysis. The significant difference was when p < 0.05.
Then, the assay was investigated step by step whether it could preserve its performance under extreme conditions. Hold time in each step including RT, initial denaturation, cycling denaturation, and annealing/extension was minimized.
Starting with RT, because the assay used WarmStart Luna Reverse Transcriptase (New England Biolabs, USA) which required incubation at 55 °C to ensure full activation, the idea of omitting the RT step by just letting the RT reaction occur while preparing the master mix at room temperature was not possible. Therefore, 5 min of RT time was compared with 4 min, 2 min, and 30 s using reference materials. Although the statistically significant difference in mean ± SEM Ct values was found, the mean difference was less than one cycle in ORF1ab (0.840) and N (0.756). Therefore, the reduction of RT time from 5 min down to 30 s very slightly affected the performance of the assay for all 3 SARS-CoV-2 gene targets. The most effect was found in ASBVd, the internal control, but the mean difference was still less than two cycles (Fig. 4A, Supplementary Table S2). Since the test was developed for use with the clinical specimen, five clinical specimens (two positive and three negative samples) were run instead of using the reference materials at RT time of 4 min and 30 s. The results revealed that 4 min was significantly better than 30 s for all 3 SARS-CoV-2 gene targets (Fig. 4B). As a result, the 4 min RT time was selected.

The performance of the adjusted real-time RT-PCR assay (A) The RT time was varied from 5 to 4 min, 2 min, and 30 s. The comparison of mean ± SEM Ct values of SARS-CoV-2 gene targets (ORF1ab, ORF3a, and N genes) and the internal control (ASBVd) was analyzed among the different RT time settings using the reference materials; (B) Comparing the mean ± SEM Ct values of SARS-CoV-2 gene targets in clinical samples at RT time between 4 min and 30 s. The significant level was calculated using an unpaired t-test and one-way ANOVA with Turkey’s multiple comparison test. The asterisk indicated the statistically significant difference. *p < 0.05, **p < 0.01, and ***p < 0.001.
In addition to denaturing the target RNA template, an initial denaturation step was required for two other purposes. First, it allowed full activation of a Hot Start Taq DNA polymerase if used. Second, it inactivated a reverse transcriptase to avoid interference with subsequent PCR steps. Since the assay used an aptamer-based Hot Start Taq DNA polymerase (New England Biolabs, USA) swiftly becoming active once the incubation temperature was above 45 °C, we speculated that the hold time of this step might also be shortened. Thus, the initial denaturation time was varied from 1 min to 30 and 2 s. The results showed that the performance of the assay using a 2 s initial denaturation was not inferior to the default 1 min setting for all three SARS-CoV-2 gene targets. (Fig. 5A, Supplementary Table S3). These findings suggested that the full function of the Hot Start Taq DNA polymerase was gained without any interference between RT and PCR when such a short hold time was used. Thus, the 2 s initial denaturation time was chosen.

The performance of the real-time RT-PCR assay. (A) when varying initial denaturation time from 1 min to 30 and 2 s; (B) when cycling denaturation and annealing/extension time was compared between 2–4 s and 1–1 s; (C) when varying annealing/extension temperatures from 60 to 65 °C; (D) when varying denaturation temperatures from 82 to 85, 88, 90, and 92 °C. The comparison of mean ± SEM Ct values of SARS-CoV-2 gene targets (ORF1ab, ORF3a, and N genes) and the internal control (ASBVd) was analyzed among the different conditions. The significant difference was calculated using an unpaired t-test and one-way ANOVA with Turkey’s multiple comparison test. The asterisk indicated the statistically significant difference. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
From previous knowledge native Taq DNA polymerase has the extension rate of 24 nucleotides per second at 55 °C and up to 45–60 nucleotides per second at 70–75°C30,31. To amplify the targets sized 76–105 bp, the annealing/extension time was supposed to be at least 2–3 s. However, considering the fact that most conventional real-time PCR instruments will spend a few seconds longer for plate read in each cycle, we hypothesized that amplification of such short products might be efficient with a 1 s annealing/extension time setting. Therefore, the cycling denaturation and annealing/extension times of 2 s and 4 s were compared with 1 s each. The results clearly showed that the performance of 1 s each was not significantly different from that of 2 s and 4 s (Fig. 5B, Supplementary Table S4). Therefore, 1 s denaturation and 1 s annealing/extension were selected.
Aside from incubation at various steps, real-time RT-PCR assay wastes substantial time ramping between denaturation and annealing/extension temperatures. Hence, the range between denaturation and annealing/extension temperatures should be reduced to shorten the run time. This could be achieved via two approaches, i.e., increment of the annealing/extension temperature and decrement of the denaturation temperature.
At first, the annealing/extension temperature was varied from 60 to 65 °C. The findings showed that the temperature could be raised to as high as 65 °C without detrimental effects on SARS-CoV-2 ORF3a and N gene targets. Meanwhile, only 60 and 61 °C worked best for the SARS-CoV-2 ORF1ab gene target and the internal control, without the delayed Ct (Fig. 5C, Supplementary Table S5).
Lastly, we predicted the melting temperature (Tm) of each amplicon using uMelt Quartz and found that the Tm of SARS-CoV-2 ORF1ab, ORF3a, and N amplicons were estimated as 84.5, 84, and 84 °C, respectively. At the same time, the predicted Tm of the ASBVd amplicon was 81.5 °C (Supplementary Fig. S1). We hypothesized that lowering the denaturation temperature down to 85 °C might be sufficient for an effective amplification. Thereby, the denaturation temperature was varied from 92 to 90, 88, 85, and 82 °C. The findings revealed that the denaturation temperature as low as 85 °C still gave an efficient amplification of all targets not different from the higher temperatures (Fig. 5D, Supplementary Fig. S2, Supplementary Table S6). Thus, denaturation at 85 °C was opted.
Altogether, the extreme setting of our newly developed assay named “Super Speed inSpector of COVID-19” or “ƩS COVID-19” was a 4 min RT step at 55 °C followed by 2 s initial denaturation at 95 °C and 40 cycles of 1 s denaturation at 85 °C and 1 s annealing/extension at 60 °C. Total run time was reduced to 24 min 46 s (more than 60% reduction compared with the standard protocol) (Fig. 6).

Time required by real-time RT-PCR (Standard protocol) and rapid real-time RT-PCR (ƩS COVID-19). The figure was created with BioRender.com.
Sensitivity of ƩS COVID-19
Analytical sensitivity of ƩS COVID-19 was determined by testing the reference SARS-CoV-2 RNA which was tenfold serially diluted to achieve 106 to 1 copies/reaction. Each concentration of the reference material was tested in 20 replicates. All agreements of the 20 replicated results were found 100% at all concentrations except at 1 copy/reaction was shown at 50% (10/20) for ORF1ab, 85% (17/20) for ORF3a, and 95% (19/20) for N. Hence, the limit of detection (LOD) from probit regression analysis of ORF1ab, ORF3a, and N genes were 1.835, 1.310, and 1 copy/reaction, respectively (Fig. 7, Supplementary Fig. S3).

The sensitivity of the rapid real-time RT-PCR assay (ƩS COVID-19). The sensitivity of ƩS COVID-19 was determined using the reference SARS-CoV-2 RNA ranging from 106 to 1 copies/reaction. Forty cycles of amplification were done in 20 replicates for each concentration of the reference RNA. The amplification plots and the standard curve of the (A) ORF1ab, (B) ORF3a, and (C) N genes of the SARS-CoV-2. The error bar represented the mean ± SEM.
Specificity of ƩS COVID-19
To determine the specificity, 22 respiratory samples positive for other 16 common respiratory viruses detected using either BioFire Respiratory 2.1 Panel or QIAstat-Dx Respiratory SARS-CoV-2 Panel were tested with ƩS COVID-19. The aforementioned respiratory viruses included coronavirus OC43, coronavirus 229E, coronavirus NL63, coronavirus HKU1, influenza A(H1N1)pdm09 virus, influenza A(H3N2) virus, influenza B virus, respiratory syncytial virus, human metapneumovirus, parainfluenza virus type 1, parainfluenza virus type 2, parainfluenza virus type 3, parainfluenza virus type 4, rhinovirus/enterovirus, adenovirus, and human bocavirus. The results confirmed that the newly developed assay had no cross-reactivity with any of these common respiratory pathogens (Fig. 8).

The specificity of the rapid real-time RT-PCR assay (ƩS COVID-19). The specificity of ƩS COVID-19 was determined using 22 respiratory samples positive for other 16 common respiratory viruses (C1-C22) detected by either BioFire Respiratory 2.1 Panel or QIAstat-Dx Respiratory SARS-CoV-2 Panel. The amplification plots of the (A) ORF1ab, (B) ORF3a, (C) N genes of SARS-CoV-2, and (D) the internal control (ASBVd).
Validation of ƩS COVID-19 in clinical samples
The newly developed assay was further validated with 83 SARS-CoV-2 positive and 100 SARS-CoV-2 negative samples determined by FDA EUA approved Cobas SARS-CoV-2 system. The results showed that ƩS COVID-19 could detect SARS-CoV-2 RNA in 77 out of 83 samples. Six discordant samples were reported as inconclusive by ƩS COVID-19, i.e., only one target gene of SARS-CoV-2 was detected (N in four samples, ORF1ab in one sample, and ORF3a in one sample). However, all these six samples were confirmed as SARS-CoV-2 detected by another FDA EUA approved assay, the Cobas SARS-CoV-2 & Influenza A/B test for use on the Cobas Liat System (Roche, Switzerland). Meanwhile, ƩS COVID-19 did not detect SARS-CoV-2 RNA in any negative samples. Therefore, the diagnostic sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and accuracy of ƩS COVID-19 were 92.8%, 100%, 100%, 97.1%, and 96.7% respectively. Almost perfected agreement (Cohen’s Kappa 0.93) was found between both assays (Table 1).